Science
Related: About this forumDiscovery of Enzymes to Catalyze Pericyclic Reactions.
A couple of days ago I attended a lecture by K.N. Houk which inspired me to look into a subject about which I have not thought for a long time, my days in synthetic chemistry being far behind me; somehow I drifted elsewhere. As a result, partly out of nostalgia, partly out of curiosity, a partly because of my disappointed realization that I could have been, should have been, more, I was inspired to check out this paper pointing to a topic about which I'd only mused casually, this many years ago: Masao Ohashi, Fang Liu, Yang Hai, Mengbin Chen, Man-cheng Tang, Zhongyue Yang, Michio Sato, Kenji Watanabe, K. N. Houk & Yi Tang, SAM-dependent enzyme-catalysed pericyclic reactions in natural product biosynthesis. Nature 549, 502–506 (2017).
The question is that while pericyclic reactions - reactions that feature, in the thermal case, 2n + 2 electrons moving in a quasi aromatic transition state (i.e., 2, 6, 10, 12...electrons, most commonly 6) in such a way that the stereochemistry is tightly controlled - are frequently used in laboratory syntheses of complex natural products, in nature, these types of reactions are not particularly well characterized in biological systems. I sort of mused about this in a half serious way; it never occurred to me to seriously look into the matter. In fact, I never even thought as to whether biological pericyclic reactions were common or even known.
It turns out they are known.
The abstract of the paper points, quite well, to the question I never more than casually asked myself:
From the paper's introduction:
A figure:
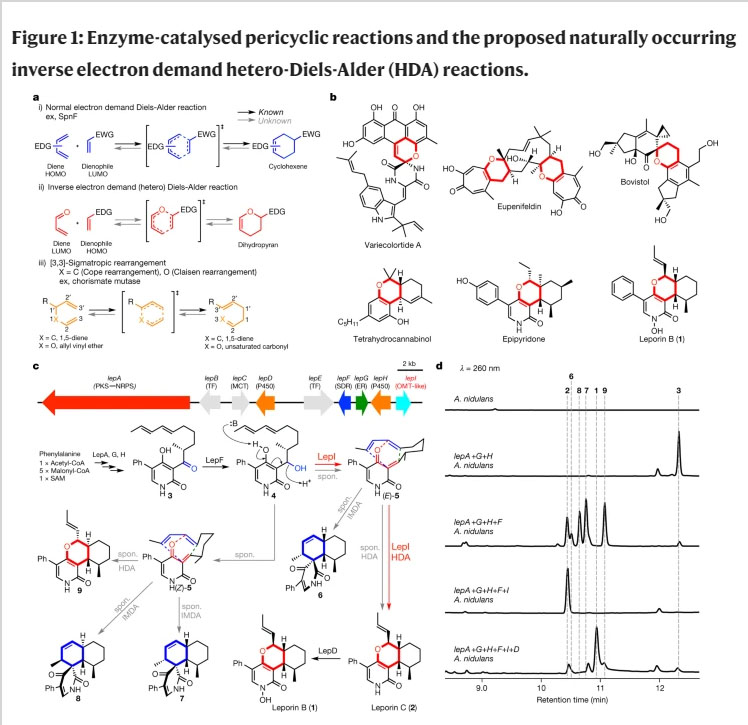
The caption:
Some more text:
The only remaining annotated enzyme in the gene cluster is LepI, which is predicted to be an O-methyltransferase (OMT) with a well-conserved SAM binding site even though no O-methylation step is required for leporin B (1) biosynthesis. When lepI was introduced into the A. nidulans strain that produced the various pericyclic products derived from reduction of 3, we were surprised to observe the exclusive production of 2 without any other products (Fig. 1d). Further addition of the P450 lepD yielded the final product 1, thereby completing heterologous pathway reconstitution (Fig. 1c, d). To first verify the function of SDR LepF, recombinant protein was expressed from Saccharomyces cerevisiae and assayed in the presence of 3 and NADPH, which yielded a single product 4 corresponding to the reduced compound (Extended Data Fig. 1). To obtain sufficient 4 for assay with LepI, we reduced 3 with NaBH4 which gave both 4 and diastereomer 4? in a ratio of approximately 1:1 (Extended Data Figs 1, 2). Each isomer was isolated and immediately added to LepI expressed and purified from Escherichia coli. Both 4 and 4? dehydrated spontaneously in the absence of LepI and afforded a mixture of IMDA (6–8) and HDA (2 and 9) products, with 2 being a very minor product (Fig. 2a). However, when LepI was added to 4, complete conversion to 2 was accomplished in the absence of any added cofactors (Fig. 2a). In contrast, addition of LepI to 4? had only a small effect on product profile. The collective in vivo and in vitro data therefore point to LepI being solely responsible for formation of 2 starting from 4, which requires stereoselective dehydration to yield (E)-5 and subsequent HDA reaction to 2...
"SAM" is a nucleoside, adenosine, that has alkylated a methionine, "S-adenosyl methionine," a sulfonium ion. It is a moiety that is often found in biosynthetic methylations. Methionine itself is one of the 20 coded amino acids.
This paper is highly cited. A description of the mechanism and the nature of the SAM cofactor's role in the mechanism can be found here: Min Chang, Yu Zhou, Hao Wang, Zihe Liu, Yi Zhang, Yue Feng, Crystal structure of the multifunctional SAM-dependent enzyme LepI provides insights into its catalytic mechanism, Biochemical and Biophysical Research Communications, Volume 515, Issue 2, 2019, Pages 255-260, and probably elsewhere.
This is a little esoteric, but it made me think a lot outside of the little box in which I live and to reflect a bit on my life and despite much happiness, some regrets.
 = new reply since forum marked as read
Highlight:
NoneDon't highlight anything
5 newestHighlight 5 most recent replies
= new reply since forum marked as read
Highlight:
NoneDon't highlight anything
5 newestHighlight 5 most recent replies

WestMichRad
(2,198 posts)I’ve been out of synthetic organic chemistry for a long time, but still appreciate the work that goes into these discoveries.